

Multicolor re-scan super-resolution imaging of live cells
Introduction
Multicolor fluorescence microscopy, which maps the distribution of different targets in cells or tissues, has proved essential in biological studies. It enables the visualization of structural or functional interactions of macromolecules, organelles, and cells (1,2). Multicolor imaging methods, such as the sequential exposure of the sample to different excitation lights (3,4) or the separate detection of different fluorophores with bandpass filters (5), have been instrumental in conventional fluorescence microscopy systems for live cell imaging. However, the diffraction-limited resolution of conventional microscopy restricts its application to imaging subcellular organelles.
Recent developments in super-resolution imaging methods, such as stimulated emission depletion microscopy (STED) (6,7), stochastic optical reconstruction microscopy (STORM) (8-10), and structured illumination microscopy (SIM) (11,12), allow the observation of biological microstructures that are unresolvable by conventional fluorescence microscopy. Nonetheless, the principles of these methods intrinsically limit their application in live cell imaging. For example, STED usually uses high laser intensity for saturated depletion [e.g., ~MW/cm2 (13)], which would severely affect the viability of live cells. STORM requires several thousand activation cycles to reconstruct a super-resolved image. Therefore, it is striking to keep the imaging sample motionless during STORM capturing, restricting its application in live cells (8). Developing strategies that can improve the resolution of the multicolor imaging of live cells is thus of great importance.
Re-scan confocal microscopy (RCM), an implementation of SIM, is a simple yet versatile super-resolution imaging method (14). It can provide at best a 2-fold improvement in lateral resolution by combining information from both excitation and emission point-spread functions (15,16). Owing to its efficient use of excitation photons, the total exposure dose to the fluorophores is significantly lower than in STED and STORM. Therefore, RCM is more suitable for live cell imaging. Furthermore, RCM possesses the advantages of confocal microscopy in live cell imaging since it can be easily achieved by simply modifying a standard confocal microscope. Therefore, it is desirable to develop a multicolor super-resolution imaging system based on an RCM setup.
A recent report presented a two-color RCM imaging method that sequentially exposes samples to two excitation lights and detects fluorescent signals separately (16,17). This method has the advantage of a simple optical path and limited crosstalk between different fluorophores due to the separate imaging process. However, the switching of laser light could lead to spatial mismatch in the resulting composite image, especially in imaging fast dynamic events. Moreover, the limited number of colors, which is determined by the number of excitation lasers, restricts the utility for multitarget studies. The simultaneous excitation and identification of multiple fluorophores in super-resolution imaging are of great practical interests for biomedical research.
In this study, we propose a novel multicolor RCM (MC-RCM) system for live cell imaging. A spectrograph is used as the multicolor detection system, and an algorithm is developed to register the intensity and spectral images. In our system, a multicolor image is formed by a single scan, which can almost entirely eliminate the spatial mismatch relative to previous studies. By imaging fluorescent beads and live cell organelles, we demonstrate that this system allows multicolor super-resolution imaging with lateral resolution up to 171 nm.
Methods
Optical system
Figure 1 shows the experimental setup. The excitation light from a 532 nm laser (SDL-532-LN-100T, Shanghai Dream Lasers Technology) was expanded and then directed to a galvanometer mirror (GM1, 6210H, Cambridge Technologies), which was used to scan the sample plane with a illumination spot to form a two-dimensional image. After the galvanometer mirror, the light was conducted through another beam expander consisting of a scan lens (SL1, CLS-SL, Thorlabs) and a tube lens (ITL200, Thorlabs) to slightly overfill the back aperture of an objective (OBJ1, UPLSAPO 60x W, 1.2 NA, Olympus). The epifluorescence from the sample was collected by the same objective, de-scanned by the GM1, passed through the dichroic mirror (DM, ZT532rdc, Chroma Technology), and focused onto a pinhole aperture by a 10×, 0.25 NA objective (OBJ2, MPLN, Olympus). The light that passed through the pinhole was collected with another 10×, 0.25 NA objective (OBJ3, MPLN, Olympus) and passed through a 550 nm longpass filter (550LP, Chroma Technology) to further remove the residual excitation light. After the longpass filter, a 30:70 beam splitter (BS, 21009, Chroma Technology) was used to divide the fluorescence signal into two parts. The thirty percent signal was re-scanned by the second galvanometer mirror (GM2, 6210H, Cambridge Technologies) and focused on a CCD camera (CoolSNAP MYO, Photometrics) for re-scan imaging, whereas the seventy percent signal was directed into a spectrograph (SP2300, PIXIS-400B, Princeton Instrument) by a 20×, 0.4 NA objective (OBJ4, MPLN, Olympus) for spectral imaging. The two galvanometer mirrors (GM1 and GM2) were controlled by a multichannel DAQ card (PCIe-6323, National Instruments). To obtain the super-resolution image, the scanning rate of GM2 was set to twice that of GM1. To obtain the wide-field image, the scanning speeds of the two galvanometer mirrors were set to the same value (14).

Reconstruction of the multicolor super-resolution image
The whole procedure for MC-RCM image reconstruction can be found in Figure 2. In brief, to acquire a multicolor super-resolution image, we recorded a super-resolution RCM image by using a CCD and a spectral image by using a spectrograph. A deconvolution algorithm was performed to obtain the RCM images. A linear spectral unmixing algorithm was implemented to obtain intensity images of individual fluorophores, which were referred to as single-color spectral images. These images were then merged to form a multicolor spectral image. By combining the super-resolution RCM image with the multicolor spectral image pixel by pixel, a MC-RCM image of the fluorophores was constructed.

The deconvolution operation was based on Richardson-Lucy (RL) algorithm detailed in our previous report (18). Briefly, the main process was as follows: first, we used theoretic estimation of the system’s PSF as the initial guess of PSF; second, two similar RL deconvolution iterations were performed to estimate PSF and object; third, when the normalized adjacent mean square error (AMSE) reached its first minimum, we terminated the computation.
Linear spectral unmixing is a classic algorithm to determine the relative abundance of materials in multispectral or hyperspectral imagery (19). This algorithm is based on the assumption that the detected signal of the mixed fluorophores can be expressed as a linear combination of these contributing fluorophores:
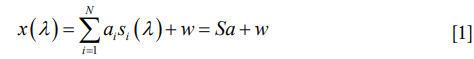
where x (λ) is the detected spectrum of the specimen which is a function of wavelength λ; N is the type number of fluorophores contained in the specimen; αi is the weight of the ith fluorophore which reflects the concertation of the fluorophore in the total signal; si (λ) is the normalized spectrum of the ith individual fluorophore (prior knowledge or measured experimentally); w is the noise. Based on the above expression, we calculated the contribution of each fluorophore using the unconstrained least squares algorithm as follows:

where ST represents a conjugate transpose of S. The single-color spectral image of an individual fluorophore can thereby be obtained by multiplying the integrated intensity of the recorded spectral image by the corresponding contribution αi pixel by pixel.
Owing to the different detection methods used in our system, the pixel numbers of the super-resolution RCM image and the spectral image are not identical. To implement a pixel-by-pixel combination, a standard bicubic interpolation was first applied to the spectral image. It should be noted that an artificial resolution improvement is introduced during the combination process because it could be considered as multiplying the RCM’s PSF by the PSF of the spectral imaging module. The product of these two PSFs is actually narrower than the PSF of the RCM. To eliminate the artificial resolution improvement, a gamma correction defined as follows was introduced to nonlinearly encode the gray values of the resulting image:

where A is a constant and usually set to 1, Vin and Vout are the pixel intensities before and after correction, respectively, and γ is the correction coefficient. In practice, 100 nm diameter fluorescent beads were imaged to measure the PSFs of both RCM and multicolor RCM. Then the γ was empirically set to 0.71, in which made the PSF of the final multicolor RCM equal to that of the RCM.
Bead sample preparation
Four kinds of fluorescent beads were employed in this study: 100 and 200 nm diameter orange fluorescent beads (540/560, F8800, F8809, Invitrogen), 200 nm diameter red fluorescent beads (580/605, F8810, Invitrogen), and 1 µm diameter crimson fluorescent beads (625/645, F8816, Invitrogen). The 100 nm diameter orange fluorescent beads mounted on the coverslip surface were used to measure the PSF of the RCM system. The other three kinds of beads were mixed and deposited on the coverslip for multicolor imaging.
Cell culture and cell sample preparation
The human breast cancer cell line SK-BR-3 and the mouse brain capillary endothelial cell line bEnd.3 were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS (Gibco) in a 37 °C incubator supplied with 5% CO2. Cells were seeded on glass bottom dishes (Nest) and allowed to adhere to the cover glass bottom overnight.
For F-actin imaging, SK-BR-3 cells were fixed with 3.7% formaldehyde for 5 minutes, then permeabilized with 0.1% Triton X-100 and finally stained with 50 µg/mL fluorescent phalloidin conjugate solution (P1951, Sigma-Aldrich) for 40 minutes at room temperature. Crucially, the cells were washed with phosphate buffered saline before the addition of another agent.
For multicolor live cell imaging, fluorescently labeled bEnd.3 cells were prepared. First, the culture medium was removed when the cells reached the desired confluence (70–80%). Then, prewarmed (37 °C) probe-containing medium was added to the dishes, and the cells were incubated under growth conditions for 2 hours. The probes were 50 nM LysoTracker Red (L7528, Thermo Fisher Scientific), which labels lysosomes, and 5 µM SYTO 82 (S11363, Thermo Fisher Scientific), which labels the nucleus. Finally, the probe-containing medium was replaced with fresh medium for imaging.
Results
To demonstrate the imaging capability of our system for resolving subcellular organelles in cells, we measured phalloidin-labeled F-actin in the breast cancer cell line SK-BR-3 (Figure 3). The images recorded by RCM (Figure 3B,E) are clearly sharper than the wild-field images (Figure 3A,D), and the deconvolution (Figure 3C,F) further improves the resolution of the RCM images. In addition, quantitative comparison of a single actin fiber was performed. As shown in Figure 3G, the FWHMs of the normalized intensity profiles of F-actin imaged in the wide-field, RCM and deconvolved RCM modes were found to be 315, 237 and 174 nm, respectively. This measurement result indicates that our system achieved a 1.8-fold resolution enhancement compared with wide-field imaging. This conclusion was further confirmed by imaging 100 nm diameter fluorescent beads. The measured FWHMs in the wide-field, RCM and deconvolved RCM modes were 305±15, 234±9 and 171±12 nm (mean ± SD, n=15 beads), respectively.

We next verified the multiple-fluorophore identification capability of our method based on the sample of mixed fluorescent beads (200 nm diameter orange fluorescent beads, 200 nm diameter red fluorescent beads, and 1 µm diameter crimson fluorescent beads). The deconvolved RCM image and the spectral image are shown in Figure 4A and B, respectively. On the basis of the spectral image (Figure 4B) and the measured spectrum of each kind of bead (Figure 4C), the three fluorophores were clearly distinguished using a linear spectral unmixing algorithm, and the corresponding single-color image was constructed (Figure 4D,E,F). By merging the three single-color images, a multicolor spectral image of the sample was obtained (Figure 4G).
To construct the MC-RCM image, we first obtained the artificial multicolor image (Figure 4H) by directly combining the deconvolved RCM image (Figure 4A) with the unmixed spectral image (Figure 4G). The artificial resolution improvement was illustrated by the observation that the diameter of a bead (indicated by double arrows in Figure 4) is smaller than that measured from the super-resolution RCM image (Figure 4A). To correct this artifact, a gamma correction with γ=0.71 was applied to the artificial multicolor image. As shown in Figure 4I, a multicolor super-resolution RCM image was eventually reconstructed, and the bead diameter was restored to approximately the value measured in the deconvolved RCM image (Figure 4A). The laser power used in this study was less than 200 µW. The imaging time for RCM was about 1 s, and the total time to obtain a multicolor RCM image was about 30 s.

Finally, we tested whether our system could image live cells. The lysosome is a subcellular organelle that is responsible for the digestion of macromolecules, old cell parts, and microorganisms. The acquisition of both lysosome and nucleus information in live cells is essential for understanding the lysosome-to-nucleus signaling mechanism (20). In this study, we imaged the mouse brain capillary endothelial cell line bEnd.3 with SYTO 82-labeled nuclei and LysoTracker Red-labeled lysosomes. The results are shown in Figure 5. Although the spectra of these two dyes largely overlap with each other (Figure 5A), we obtained the respective super-resolution images of the nuclei (Figure 5C,G) and lysosomes (Figure 5D,H) without any crosstalk. These results show that our imaging system is capable of performing multicolor super-resolution imaging of live cells and may contribute to revealing biological mechanisms at the organelle even at the molecular level, such as lysosome-to-nucleus signaling.

Discussion and conclusions
In contrast to multifocal SIM (MSIM) (16) and instant SIM (ISIM) (17), which provide a dual-color super-resolution image by scanning the sample twice with two different lasers, in this study, we proposed a new method to obtain a multicolor super-resolution image. This method was developed based on spectral imaging combined with a linear spectral unmixing process and the registration of unmixed spectral images with super-resolution RCM images. Using this method, a single scan of the sample suffices to acquire a multicolor super-resolution image. The increased imaging speed is important for monitoring rapid dynamic events involving interactions between multiple molecules and organelles. Furthermore, shortening the scanning time decreases the probability of fluorescence photobleaching, which benefits live cell imaging.
Despite the advantages mentioned above, the exposure time of 30 s for a single-frame multicolor super-resolution imaging is not fast enough to capture transient dynamics in live cells. The imaging rate of our system is restricted by the speed of the spectrograph, which is 1,300 spectra per second. Each pixel has its own spectrum and a spectral image usually has 200×200 pixels. Thus, the spectral imaging speed of our multicolor RCM system is 1,300/40,000=0.0325 fps, far below the RCM imaging rate. For example, to cover a 20×20 µm2 field of view at a 100 nm step size would take a total acquisition time of ~30 s, which is 30 times longer than the RCM imaging process itself. In the future, we will replace the spectrograph with multichannel photomultiplier tubes to improve the imaging rate.
Acknowledgments
Funding: National Key Research and Development Program of China (2017YFC0110200); Program 973 (2015CB755502); National Natural Science Foundation of China (NSFC) (81471702, 81822023, 81701744, 81571724); Natural Science Foundation of Guangdong Province (2014A030312006, 2017A030310308); The Scientific Instrument Innovation Team of Chinese Academy of Sciences (GJJSTD20180002); Shenzhen Basic Research Program (JCYJ20170818164343304, JCYJ20170818155006471, JCYJ20160608214524052); SIAT Innovation Program for Excellent Young Researchers (201821).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Bates M, Dempsey GT, Chen KH, Zhuang X. Multicolor Super-Resolution Fluorescence Imaging via Multi-Parameter Fluorophore Detection. Chemphyschem 2012;13:99-107. [Crossref] [PubMed]
- Klein T, van de Linde S, Sauer M. Live-Cell Super-Resolution Imaging Goes Multicolor. Chembiochem 2012;13:1861-3. [Crossref] [PubMed]
- Fiolka R, Shao L, Rego EH, Davidson MW, Gustafsson MGL. Time-lapse two-color 3D imaging of live cells with doubled resolution using structured illumination. Proc Natl Acad Sci U S A 2012;109:5311-5. [Crossref] [PubMed]
- Mahou P, Zimmerley M, Loulier K, Matho KS, Labroille G, Morin X, Supatto W, Livet J, Debarre D, Beaurepaire E. Multicolor two-photon tissue imaging by wavelength mixing. Nat Methods 2012;9:815-18. [Crossref] [PubMed]
- Testa I, Wurm CA, Medda R, Rothermel E, von Middendorf C, Foelling J, Jakobs S, Schoenle A, Hell SW, Eggeling C. Multicolor Fluorescence Nanoscopy in Fixed and Living Cells by Exciting Conventional Fluorophores with a Single Wavelength. Biophys J 2010;99:2686-94. [Crossref] [PubMed]
- Hell SW. Far-field optical nanoscopy. Science 2007;316:1153-8. [Crossref] [PubMed]
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 1994;19:780-2. [Crossref] [PubMed]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 2006;3:793-5. [Crossref] [PubMed]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 2007;317:1749-53. [Crossref] [PubMed]
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008;319:810-3. [Crossref] [PubMed]
- Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 2000;198:82-7. [Crossref] [PubMed]
- Gustafsson MGL. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A 2005;102:13081-6. [Crossref] [PubMed]
- Willig KI, Harke B, Medda R, Hell SW. STED microscopy with continuous wave beams. Nat Methods 2007;4:915-8. [Crossref] [PubMed]
- De Luca GM, Breedijk RM, Brandt RA, Zeelenberg CH, de Jong BE, Timmermans W, Azar LN, Hoebe RA, Stallinga S, Manders EM. Re-scan confocal microscopy: scanning twice for better resolution. Biomed Opt Express 2013;4:2644-56. [Crossref] [PubMed]
- De Luca G, Breedijk R, Hoebe R, Stallinga S, Manders E. Re-scan confocal microscopy (RCM) improves the resolution of confocal microscopy and increases the sensitivity. Methods Appl Fluoresc 2017;5:015002. [Crossref] [PubMed]
- York AG, Parekh SH, Nogare DD, Fischer RS, Temprine K, Mione M, Chitnis AB, Combs CA, Shroff H. Resolution doubling in live, multicellular organisms via multifocal structured illumination microscopy. Nat Methods 2012;9:749-54. [Crossref] [PubMed]
- York AG, Chandris P, Nogare DD, Head J, Wawrzusin P, Fischer RS, Chitnis A, Shroff H. Instant super-resolution imaging in live cells and embryos via analog image processing. Nat Methods 2013;10:1122-6. [Crossref] [PubMed]
- Chen J, Lin R, Wang H, Meng J, Zheng H, Song L. Blind-deconvolution optical-resolution photoacoustic microscopy in vivo. Opt Express 2013;21:7316-27. [Crossref] [PubMed]
- Keshava N, Mustard JF. Spectral unmixing. IEEE Signal Processing Magazine 2002;19:44-57. [Crossref]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Tuong H, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012;31:1095-108. [Crossref] [PubMed]

